
The American Society of Plastic Surgeons reports that demand for minimally invasive cosmetic procedures has grown significantly over the past decade, reflecting a broader interest in appearance-related wellness and preventive self-care. This growth has expanded the aesthetic industry beyond traditional cosmetic treatments, creating a marketplace where consumers are presented with a wide range of providers, technologies, and service models.
As interest in aesthetic treatments increases, patients often spend considerable time researching clinics before scheduling appointments. Many consumers review educational resources, provider credentials, patient feedback, and examples of award-winning care in Ashburn VA as part of their broader evaluation process. While recognition and branding may attract attention, informed decision-making typically involves a much deeper assessment of quality, safety, and overall patient experience.
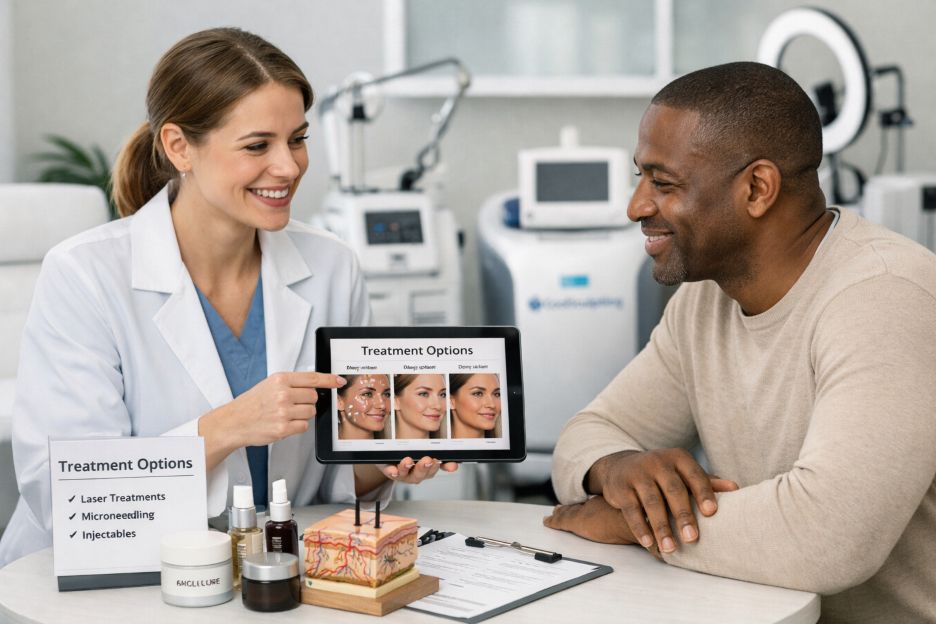
Rising Demand for Aesthetic and Wellness Services
Aesthetic medicine has evolved considerably in recent years. Procedures once associated primarily with cosmetic enhancement are increasingly discussed within larger wellness conversations. Treatments focused on skin health, rejuvenation, preventive aging, and personal confidence now appeal to a diverse range of age groups.
Research published by the International Society of Aesthetic Plastic Surgery (ISAPS) indicates continued growth in both surgical and non-surgical procedures worldwide. Factors contributing to this trend include increased public awareness, advances in technology, shorter recovery times, and broader access to treatment options.
Consumers today are generally more informed than previous generations. Online reviews, professional directories, educational articles, and social media platforms provide access to information that allows patients to compare providers before making decisions. However, the abundance of information can also make provider selection more complex.
Factors Patients Evaluate When Selecting Providers
When considering aesthetic treatments, consumers often evaluate multiple factors simultaneously rather than focusing on a single characteristic.
One of the most important considerations is the provider’s qualifications. Patients commonly seek professionals with relevant medical training, certifications, and experience performing specific procedures. Specialized education helps demonstrate that providers possess the knowledge required to assess risks, recommend appropriate treatments, and respond to potential complications.
Facility standards also influence decision-making. A clean, organized environment can communicate professionalism and attention to patient safety. Many consumers pay close attention to the appearance of treatment rooms, sanitation practices, and overall clinic operations during initial visits.
Technology is another area of interest. While advanced equipment may offer certain advantages, experts note that technology should be viewed as one component of care rather than the sole indicator of quality. Effective outcomes depend on appropriate patient selection, proper technique, and realistic treatment planning.
Cost remains a consideration as well. However, patients increasingly recognize that the lowest price does not always represent the best value. Many consumers compare treatment plans, consultation quality, provider expertise, and safety standards alongside pricing information.
The Importance of Credentials, Safety Protocols, and Consultation Processes
Credentials often serve as an important starting point when evaluating providers. Medical licensing, professional memberships, continuing education, and specialized certifications can help demonstrate commitment to professional standards.
The U.S. Food and Drug Administration (FDA) emphasizes that aesthetic treatments involve medical considerations and should be approached with appropriate attention to safety. Consumers are encouraged to verify qualifications and understand the benefits and risks associated with any procedure they are considering.
Safety protocols are equally important. Reputable clinics typically maintain clear procedures related to patient screening, infection prevention, equipment maintenance, informed consent, and emergency preparedness. These systems may not be immediately visible to patients, but they play a critical role in protecting health and supporting treatment outcomes.
The consultation process often reveals valuable information about a clinic’s approach to care. High-quality consultations generally involve:
- A thorough discussion of patient goals and concerns.
- Review of medical history and relevant health factors.
- Explanation of treatment options and alternatives.
- Discussion of potential risks and expected outcomes.
- Clear guidance regarding recovery and follow-up care.
Experts from the American Academy of Dermatology note that realistic expectations are essential for patient satisfaction. Consultations that prioritize education rather than sales pressure often help patients make more informed decisions.
How Reputation and Patient Experience Influence Decision-Making
Reputation remains a significant factor in healthcare-related decisions. Online reviews, referrals from friends and family, and testimonials frequently shape consumer perceptions before an appointment is ever scheduled.
However, patient experience extends far beyond treatment results. Many consumers evaluate the entire journey, including appointment scheduling, communication quality, staff professionalism, waiting times, and post-treatment support.
Research published by the Beryl Institute, an organization focused on patient experience improvement, suggests that individuals often associate positive healthcare experiences with clear communication, respect, empathy, and responsiveness. These factors can influence overall satisfaction even when clinical outcomes are similar.
Trust also plays an important role. Patients frequently seek providers who listen carefully, answer questions honestly, and present balanced information about both benefits and limitations. Transparency can strengthen confidence and support long-term provider relationships.
Understanding Industry Recognition and Professional Reputation
The concept of excellence in aesthetic care is often reflected through awards, certifications, peer recognition, and positive patient feedback. While these indicators can provide useful information, consumers should evaluate them within a broader context.
Industry awards may highlight customer service, innovation, or community reputation. However, awards alone do not necessarily guarantee clinical quality. Similarly, positive online reviews can provide insight into patient experiences, but individual reviews should be considered alongside other objective factors.
Consumers benefit from examining multiple indicators simultaneously. Professional credentials, consultation quality, safety standards, treatment transparency, and patient education often provide a more complete picture than any single form of recognition.
Within the broader aesthetic industry, the discussion surrounding recognized providers and highly rated wellness facilities illustrates an important principle: meaningful quality assessment requires a combination of objective verification and personal evaluation.
Practical Guidance for Evaluating Treatment Providers Objectively
Consumers can improve their decision-making process by approaching provider selection systematically.
- Verify professional licenses and certifications.
- Research experience related to specific treatments.
- Schedule consultations with more than one provider when appropriate.
- Ask questions about safety protocols and follow-up care.
- Review patient feedback while maintaining realistic expectations.
- Consider communication style and overall comfort level.
- Evaluate treatment recommendations carefully rather than making decisions based solely on promotions or pricing.
Taking time to gather information can help patients distinguish between marketing claims and evidence-based care. Objective evaluation becomes especially important when considering procedures that involve significant financial investment or potential health risks. For individuals exploring surgical options, resources that explain how to choose a cosmetic surgeon can provide additional guidance on evaluating qualifications, experience, consultation practices, and overall treatment suitability.
Conclusion
The growing popularity of aesthetic and wellness services has created more choices for consumers than ever before. While advanced technology, positive reviews, and professional recognition may influence initial impressions, long-term satisfaction often depends on deeper factors such as provider qualifications, safety practices, consultation quality, and patient-centered care.
Consumers who approach provider selection thoughtfully are better positioned to make informed decisions that align with their goals and expectations. By focusing on credentials, transparent communication, established safety standards, and overall treatment experience, individuals can evaluate aesthetic clinics more objectively and navigate the evolving beauty and wellness landscape with greater confidence.



 Eyelid surgery is performed to improve the overall look of eyes by removing excess tissues like skin, fat, and muscle from the upper and lower eyelids. It helps to create a more youthful & attractive appearance. While this surgery can be a great way to improve the look of your eyes, it’s important to take some pre-care steps to ensure a successful outcome. Here are some essential things you need to do before undergoing eyelid surgery.
Eyelid surgery is performed to improve the overall look of eyes by removing excess tissues like skin, fat, and muscle from the upper and lower eyelids. It helps to create a more youthful & attractive appearance. While this surgery can be a great way to improve the look of your eyes, it’s important to take some pre-care steps to ensure a successful outcome. Here are some essential things you need to do before undergoing eyelid surgery. There are plenty of good reasons why we need to be at a healthy weight and maintain it. Know these facts. An individual with a Body Mass Index (BMI) that ranges from 30 to 35 may die 2 to 4 years earlier compared to a person with
There are plenty of good reasons why we need to be at a healthy weight and maintain it. Know these facts. An individual with a Body Mass Index (BMI) that ranges from 30 to 35 may die 2 to 4 years earlier compared to a person with  Exipure is a dietary supplement for weight loss made from natural ingredients which has a unique system of action targeting the processes of burning fat as well as improving them. This weight loss formula makes use of natural extracts from eight herbs and plants blended in appropriate amounts. This blend of extracts increases the brown adipose tissue (BAT) levels in the body so as to make weight loss possible.
Exipure is a dietary supplement for weight loss made from natural ingredients which has a unique system of action targeting the processes of burning fat as well as improving them. This weight loss formula makes use of natural extracts from eight herbs and plants blended in appropriate amounts. This blend of extracts increases the brown adipose tissue (BAT) levels in the body so as to make weight loss possible.
 The facial skin is the body’s region, and some other anomalies in signs or attributes of aging may be seen. But a selection of decorative cosmetic surgery processes is available now to supply a young and appealing facial look.
The facial skin is the body’s region, and some other anomalies in signs or attributes of aging may be seen. But a selection of decorative cosmetic surgery processes is available now to supply a young and appealing facial look. People today undergo cosmetic surgery for a lot of factors. Some wish to appear younger. Other people attempt to alter a characteristic they have never enjoyed. The choice is private. Among these secrets is to establish realistic expectations. Cosmetic Surgery will not alter your own life. It will not solve personal issues or cause you to seem like somebody else. But it might provide you higher self-confidence and enhance your feeling of well-being. Successful outcomes often rely, in part, on how well you and your physician convey. Be certain that you feel comfortable with your physician and that you’re available for him or her about your objectives and questions.
People today undergo cosmetic surgery for a lot of factors. Some wish to appear younger. Other people attempt to alter a characteristic they have never enjoyed. The choice is private. Among these secrets is to establish realistic expectations. Cosmetic Surgery will not alter your own life. It will not solve personal issues or cause you to seem like somebody else. But it might provide you higher self-confidence and enhance your feeling of well-being. Successful outcomes often rely, in part, on how well you and your physician convey. Be certain that you feel comfortable with your physician and that you’re available for him or her about your objectives and questions.
 What’s Cosmetic Surgery?
What’s Cosmetic Surgery? Cosmetic surgery is an area of science connected with adjusting a human body part’s shape and functioning. Most operational and deformities deficiencies could be completely or partly removed with the support of this science fiction. The Greek term “Plastikos” gets the root of the process which means match to mold.
Cosmetic surgery is an area of science connected with adjusting a human body part’s shape and functioning. Most operational and deformities deficiencies could be completely or partly removed with the support of this science fiction. The Greek term “Plastikos” gets the root of the process which means match to mold.
 Botox is a cosmetic treatment or surgery to tighten loose skin. This is a treatment often used by Hollywood stars to prevent the signs of ageing from appearing.
Botox is a cosmetic treatment or surgery to tighten loose skin. This is a treatment often used by Hollywood stars to prevent the signs of ageing from appearing. The average person if asked what plastic surgery is, they would answer Botox and Fillers. Plastic surgery covers head to toe procedures, from bones to blood vessels, from muscles to nerves. Plastic surgery has a dual value. One is that it solves a problem on open wound or a deficient part and the second aspect of it is the emotional reaction of the person who has that problem. Plastic surgery can usually help 90% of the person walking in clinics of plastic surgeons. It is a field of medicine that very few surgeons have ever regretted going into.
The average person if asked what plastic surgery is, they would answer Botox and Fillers. Plastic surgery covers head to toe procedures, from bones to blood vessels, from muscles to nerves. Plastic surgery has a dual value. One is that it solves a problem on open wound or a deficient part and the second aspect of it is the emotional reaction of the person who has that problem. Plastic surgery can usually help 90% of the person walking in clinics of plastic surgeons. It is a field of medicine that very few surgeons have ever regretted going into.
 Loss of blood is expected uncontrolled and large loss of blood can lead to a fall in blood pressure and may be dangerous With any surgery.
Loss of blood is expected uncontrolled and large loss of blood can lead to a fall in blood pressure and may be dangerous With any surgery.







 Optimal liver function helps to metabolize fat and detoxify the body. The liver responds to the metabolism processes by releasing bile that works with an enzyme called Lipase.
Optimal liver function helps to metabolize fat and detoxify the body. The liver responds to the metabolism processes by releasing bile that works with an enzyme called Lipase. Consumers who read the latest Consumer Reports finally understood how Liv Pure works in improving the
Consumers who read the latest Consumer Reports finally understood how Liv Pure works in improving the 

 As you get older, it’s essential to make sure your body gets the right nutrition. Taking natural health supplements like Hildegard Maitrunk can help you achieve this and make sure your body receives all the essential vitamins, minerals and other nutrients
As you get older, it’s essential to make sure your body gets the right nutrition. Taking natural health supplements like Hildegard Maitrunk can help you achieve this and make sure your body receives all the essential vitamins, minerals and other nutrients 


 When considering using human growth hormones as a means of increasing muscle mass, it’s important to know there is little research supporting its effectiveness. While it is true that human growth hormone (HGH) therapy is recommended for young patients diagnosed as afflicted with growth hormone deficiency, it doesn’t mean that the therapy will produce the same results in healthy adults.
When considering using human growth hormones as a means of increasing muscle mass, it’s important to know there is little research supporting its effectiveness. While it is true that human growth hormone (HGH) therapy is recommended for young patients diagnosed as afflicted with growth hormone deficiency, it doesn’t mean that the therapy will produce the same results in healthy adults. In the case of HGH, these are hormones that signal the liver to release a substance called insulin-like growth factor 1, a.k.a. IGF-1. This is the substance that directly helps patients with liver disorder, to help the body build muscle mass.
In the case of HGH, these are hormones that signal the liver to release a substance called insulin-like growth factor 1, a.k.a. IGF-1. This is the substance that directly helps patients with liver disorder, to help the body build muscle mass. Researchers explain that inasmuch as there is increased
Researchers explain that inasmuch as there is increased 
 In the case of Magnesium, supplements chemists used the amino acid taurine as a chelating agent in producing a more stable and nutritive compound called Magnesium Taurate.
In the case of Magnesium, supplements chemists used the amino acid taurine as a chelating agent in producing a more stable and nutritive compound called Magnesium Taurate.


